Une récente étude parue dans la revue Science le 17 avril 2024 ( https://doi.org/10.1126/sciadv.adi8419 ) effectuée à l’Université de Yokohama précise l’origine de la population du Japon moderne. L’occupation de l’archipel débuta vers 14000 ans avant l’ère commune par des chasseurs-cueilleurs de l’ère Han provenant de la région du Fleuve Jaune aujourd’hui en Chine. Cette occupation donna lieu à la culture Jomon qui malgré le fait qu’elle est classée dans la période du néolithique fut caractérisée par la production de poteries considérées comme les plus anciennes dans le monde (Musée national du Japon).

Cette immigration fut suivie d’une autre vague humaine qui s’établit dans l’archipel d’Okinawa et qui se mélangea considérablement avec le peuple Jomon. Il subsiste une incertitude quant à l’origine de ce peuple qui arriva Vers 500 à 300 avant l’ère commune. Dans l’île de Miyako de cet archipel les caractéristiques génétiques sont les plus marquées, encore aujourd’hui. Cette origine génétique n’a pas donné lieu à des études détaillées alors que l’étude présente a procédé à une analyse très détaillée concernant 3256 génomes équitablement répartis sur tout l’archipel nippon.

Puis une troisième vague d ‘« invasions » provenant de la péninsule de Corée et constituée d’un mélange génétique entre Coréens et Chinois Han des régions chinoises au nord du Fleuve Jaune, mélange appelé admixture, débuta vers le début de l’ère commune. Apparemment la proximité de l’île de Taïwan avec l’archipel d’Okinawa n’a pas été favorable à ces vagues d’immigration à moins que les aborigènes encore présents dans cette île ne soient pas différenciables des habitants du nord du Fleuve Jaune. Cette étude a en outre montré une présence non négligeable de gènes Denisovans et de gènes d’origine néandertalienne. Ces gènes denisovans se retrouvent distinctement dans l’île Ayta des Philippines et les gènes concernés par des origines néandertaliennes se retrouvent notamment en Islande ! Comme dans le cas des populations présente dans la région asiatique de l’est la culture du riz n’a été introduite au Japon que durant l’ère Yayoi qui succéda à l’ère Jomon vers 300 avant l’ère commune. Il est vrai que la culture du riz est sophistiquée dans la mesure où il est nécessaire de préparer des terres arables parfaitement plates, de disposer d’eau pour contrôler leur inondation et enfin produire des pousses de riz. L’origine de culture du riz est également chinoise.
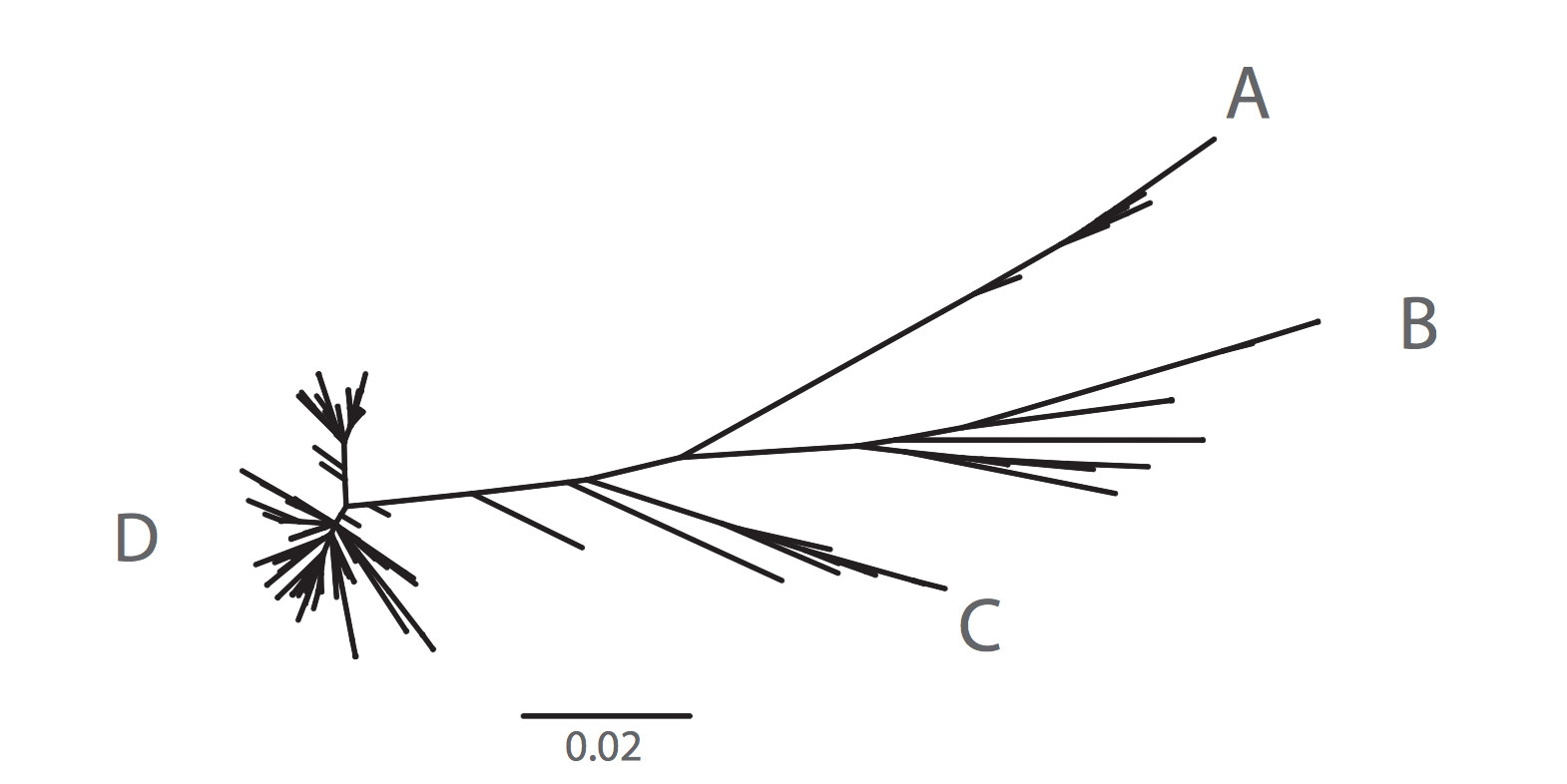
Afin de comprendre l’illustration provenant de l’article cité en référence il est nécessaire de connaître en quoi consiste une analyse par composant principal d’une variable (voir la note en fin d’article). Cependant les analyses effectuées dans le cas de la génétique du peuple du Japon par cette technique a montré que l’extension des peuples ayant envahi le Japon par vagues successives depuis la péninsule coréenne dans tout l’archipel jusqu’à Hokkaido fut un long processus qui dura près de 15000 ans avant l’ère commune. Il n’est mentionné nulle part dans cette étude la présence d’immigration en provenance de la Sibérie orientale car il s’agit probablement de vagues humaines issues à nouveau de la région du Fleuve Jaune dont il faut rappeler ici qu’elle fut le berceau de nombreuses inventions technologiques alors que les civilisations grecque et assyrienne n’existaient tout simplement pas ! Il est probable que le savoir-faire Jomon pour la confection de poteries datant de plus de 10000 ans avant l’ère commune est issu de cette région de la Chine. Il est raisonnable de conclure que la civilisation japonaise fait également partie d’une des plus vieilles du monde.

Note explicative. PCA = analyse du paramètre principal, AL = fréquence des allèles, MAF = fréquence des allèles mineurs, PCA-UMAP = outil d’approximation et de projection pour analyse du paramètre principal (PCA), K facteur de contrainte de l’analyse par composant principal par région du Japon.